Benefits for scientists
- More detailed and deeper understanding of underlying mechanisms that are often beyond experimental reach.
- Validation of governing equations by comparison with experimental data.
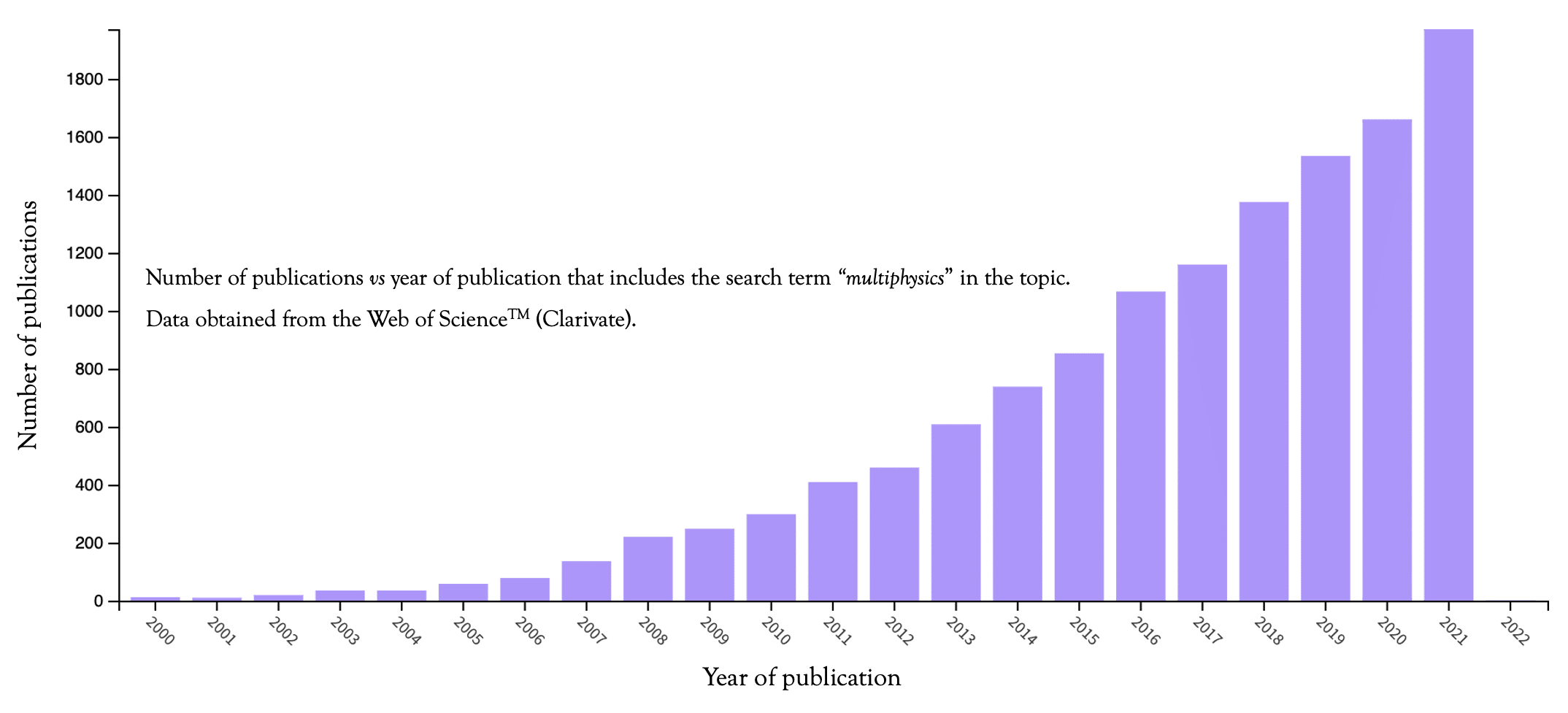
- The number of scientific publications incorporating multiphysics modeling has increased exponentially year after year.
- Possessing good multiphysics modeling skills increases the quality and quantity of research papers, which has a direct impact on the success of a scientific career.
Benefits for engineers
- Generation of highly realistic digital twins.
- Accuracy and optimization of all types of designs, products and devices.
- Very interesting to add to the catalog of skills and knowledge of today’s engineers.
- Increases the versatility and efficiency of design and optimization tasks, a fact that is obviously highly valued by technological and industrial companies.
Benefits for companies
- It enables R&D departments to create complex models of their systems and devices.
- Product and process performance can be optimized and potential problems can be solved, saving time, work and subsequent costs.
- Training in multiphysics modeling is essential for the efficiency of the R&D process in technology and industrial companies, which will ultimately result in improved company competitiveness.